
A study in GenomeBiology finds that protein length distribution is a remarkably consistent feature across species. This is in contrast to other genomic features, suggesting that protein length may be subject to unique evolutionary constraints.
]. In this context, the length distribution of proteins could be the result of an optimization process whereby adding new domains may contribute to functional flexibility, albeit at an energy cost for the cell, with diminishing returns.
], making them easier to regulate than longer genes. A full exploration of these hypotheses is beyond the scope of this article.Our comprehensive survey of 2326 species has demonstrated that protein length distribution is a remarkably consistent feature across species. This finding stands in stark contrast to other genomic features and suggests that protein length may be subject to unique evolutionary constraints.
Moving forward, our results invoke intriguing questions about the underlying mechanisms shaping gene repertoire evolution, and future studies will be needed to explore the causes of this unexpected consistency.Data regarding genomic features of all species were extracted from the August 2020 release of the OMA Database []. It consists of 2326 species: 485 eukaryotes, 153 archaea, and 1688 bacteria.
: Genome length data is not available in OMA and not easily obtainable due to the heterogeneity of different data sources. We estimated the genome size by adding for each chromosome or contig, the difference between the 3′-most position of the 3′-most genes and the 5′-most position of the 5′-most gene. This is an estimate that systemically underestimates the real genome length, but is likely to be of a similar order of magnitude.
], as the isoform with the highest sequence match compared to orthologous sequences across all species. For each gene, the values for the gene-centric metrics were obtained as follows:: The length of the string representing the amino-acid sequence of the protein stored in OMA.: The difference between the 3′-most position of the gene and the 5′-most position of the gene, as sorted in OMA. These positions account for untranslated regions.
Belgique Dernières Nouvelles, Belgique Actualités
Similar News:Vous pouvez également lire des articles d'actualité similaires à celui-ci que nous avons collectés auprès d'autres sources d'information.
 mutscan—a flexible R package for efficient end-to-end analysis of multiplexed assays of variant effect data - Genome BiologyMultiplexed assays of variant effect (MAVE) experimentally measure the effect of large numbers of sequence variants by selective enrichment of sequences with desirable properties followed by quantification by sequencing. mutscan is an R package for flexible analysis of such experiments, covering the entire workflow from raw reads up to statistical analysis and visualization. The core components are implemented in C++ for efficiency. Various experimental designs are supported, including single or paired reads with optional unique molecular identifiers. To find variants with changed relative abundance, mutscan employs established statistical models provided in the edgeR and limma packages. mutscan is available from https://github.com/fmicompbio/mutscan .
mutscan—a flexible R package for efficient end-to-end analysis of multiplexed assays of variant effect data - Genome BiologyMultiplexed assays of variant effect (MAVE) experimentally measure the effect of large numbers of sequence variants by selective enrichment of sequences with desirable properties followed by quantification by sequencing. mutscan is an R package for flexible analysis of such experiments, covering the entire workflow from raw reads up to statistical analysis and visualization. The core components are implemented in C++ for efficiency. Various experimental designs are supported, including single or paired reads with optional unique molecular identifiers. To find variants with changed relative abundance, mutscan employs established statistical models provided in the edgeR and limma packages. mutscan is available from https://github.com/fmicompbio/mutscan .
Lire la suite »
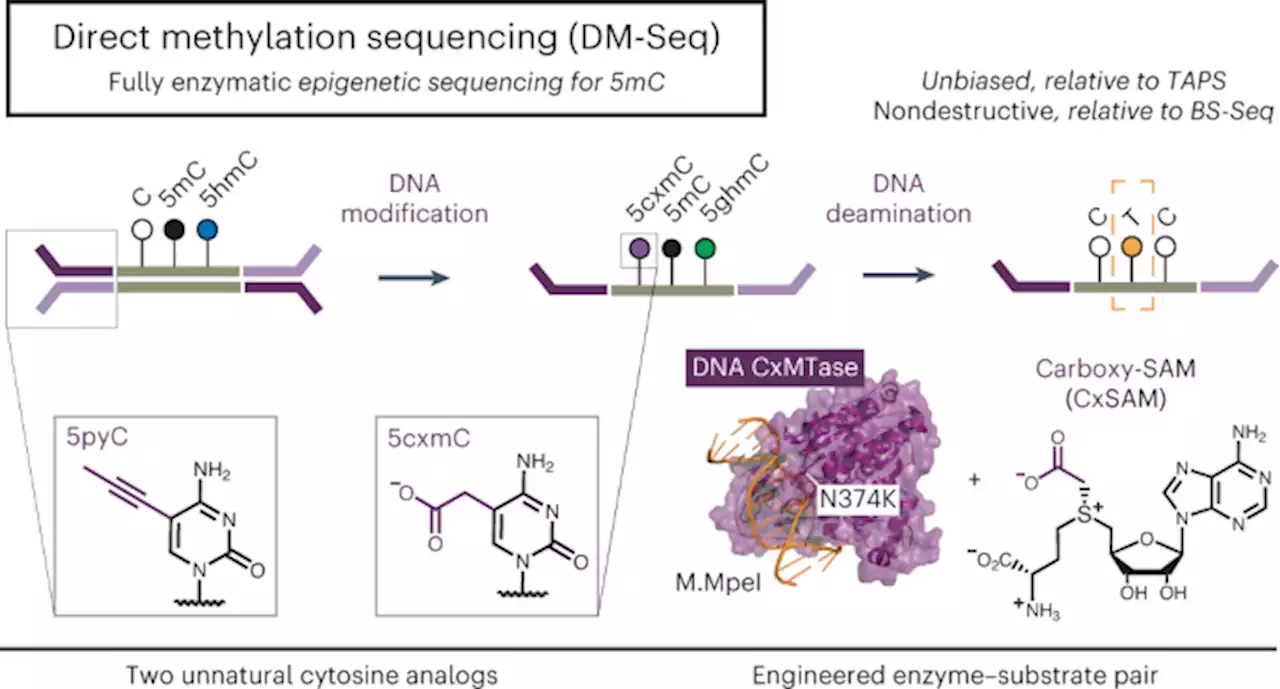 Direct enzymatic sequencing of 5-methylcytosine at single-base resolution - Nature Chemical BiologyWang et al. developed a bisulfite-free method termed DM-Seq that leverages an unnatural enzyme–substrate pair coupled with a DNA deaminase to sequence 5-methylcytosine at base resolution in sparse DNA samples, circumventing the limitations of chemical deamination methods.
Direct enzymatic sequencing of 5-methylcytosine at single-base resolution - Nature Chemical BiologyWang et al. developed a bisulfite-free method termed DM-Seq that leverages an unnatural enzyme–substrate pair coupled with a DNA deaminase to sequence 5-methylcytosine at base resolution in sparse DNA samples, circumventing the limitations of chemical deamination methods.
Lire la suite »
 A new weapon against COVID: RBD-62 protein shows promising results in preventing severe diseaseA new weapon against COVID: RBD-62 protein shows promising results in preventing severe disease biorxivpreprint covid COVID SARSCoV2 disease immunology disease severity
A new weapon against COVID: RBD-62 protein shows promising results in preventing severe diseaseA new weapon against COVID: RBD-62 protein shows promising results in preventing severe disease biorxivpreprint covid COVID SARSCoV2 disease immunology disease severity
Lire la suite »
 RELA governs a network of islet-specific metabolic genes necessary for beta cell function - DiabetologiaAims/hypothesis NF-κB activation unites metabolic and inflammatory responses in many diseases yet less is known about the role that NF-κB plays in normal metabolism. In this study we investigated how RELA impacts the beta cell transcriptional landscape and provides network control over glucoregulation. Methods We generated novel mouse lines harbouring beta cell-specific deletion of either the Rela gene, encoding the canonical NF-κB transcription factor p65 (βp65KO mice), or the Ikbkg gene, encoding the NF-κB essential modulator NEMO (βNEMOKO mice), as well as βA20Tg mice that carry beta cell-specific and forced transgenic expression of the NF-κB-negative regulator gene Tnfaip3, which encodes the A20 protein. Mouse studies were complemented by bioinformatics analysis of human islet chromatin accessibility (assay for transposase-accessible chromatin with sequencing [ATAC-seq]), promoter capture Hi-C (pcHi-C) and p65 binding (chromatin immunoprecipitation–sequencing [ChIP-seq]) data to investigate genome-wide control of the human beta cell metabolic programme. Results Rela deficiency resulted in complete loss of stimulus-dependent inflammatory gene upregulation, consistent with its known role in governing inflammation. However, Rela deletion also rendered mice glucose intolerant because of functional loss of insulin secretion. Glucose intolerance was intrinsic to beta cells as βp65KO islets failed to secrete insulin ex vivo in response to a glucose challenge and were unable to restore metabolic control when transplanted into secondary chemical-induced hyperglycaemic recipients. Maintenance of glucose tolerance required Rela but was independent of classical NF-κB inflammatory cascades, as blocking NF-κB signalling in vivo by beta cell knockout of Ikbkg (NEMO), or beta cell overexpression of Tnfaip3 (A20), did not cause severe glucose intolerance. Thus, basal p65 activity has an essential and islet-intrinsic role in maintaining normal glucose homeostasis. Genome-wide bio
RELA governs a network of islet-specific metabolic genes necessary for beta cell function - DiabetologiaAims/hypothesis NF-κB activation unites metabolic and inflammatory responses in many diseases yet less is known about the role that NF-κB plays in normal metabolism. In this study we investigated how RELA impacts the beta cell transcriptional landscape and provides network control over glucoregulation. Methods We generated novel mouse lines harbouring beta cell-specific deletion of either the Rela gene, encoding the canonical NF-κB transcription factor p65 (βp65KO mice), or the Ikbkg gene, encoding the NF-κB essential modulator NEMO (βNEMOKO mice), as well as βA20Tg mice that carry beta cell-specific and forced transgenic expression of the NF-κB-negative regulator gene Tnfaip3, which encodes the A20 protein. Mouse studies were complemented by bioinformatics analysis of human islet chromatin accessibility (assay for transposase-accessible chromatin with sequencing [ATAC-seq]), promoter capture Hi-C (pcHi-C) and p65 binding (chromatin immunoprecipitation–sequencing [ChIP-seq]) data to investigate genome-wide control of the human beta cell metabolic programme. Results Rela deficiency resulted in complete loss of stimulus-dependent inflammatory gene upregulation, consistent with its known role in governing inflammation. However, Rela deletion also rendered mice glucose intolerant because of functional loss of insulin secretion. Glucose intolerance was intrinsic to beta cells as βp65KO islets failed to secrete insulin ex vivo in response to a glucose challenge and were unable to restore metabolic control when transplanted into secondary chemical-induced hyperglycaemic recipients. Maintenance of glucose tolerance required Rela but was independent of classical NF-κB inflammatory cascades, as blocking NF-κB signalling in vivo by beta cell knockout of Ikbkg (NEMO), or beta cell overexpression of Tnfaip3 (A20), did not cause severe glucose intolerance. Thus, basal p65 activity has an essential and islet-intrinsic role in maintaining normal glucose homeostasis. Genome-wide bio
Lire la suite »
 Genome-wide mapping of gene-microbe interactions in the murine lung microbiota based on quantitative microbial profiling - Animal MicrobiomeBackground Mammalian lungs comprise a complex microbial ecosystem that interacts with host physiology. Previous research demonstrates that the environment significantly contributes to bacterial community structure in the upper and lower respiratory tract. However, the influence of host genetics on the makeup of lung microbiota remains ambiguous, largely due to technical difficulties related to sampling, as well as challenges inherent to investigating low biomass communities. Thus, innovative approaches are warranted to clarify host-microbe interactions in the mammalian lung. Results Here, we aimed to characterize host genomic regions associated with lung bacterial traits in an advanced intercross mouse line (AIL). By performing quantitative microbial profiling (QMP) using the highly precise method of droplet digital PCR (ddPCR), we refined 16S rRNA gene amplicon-based traits to identify and map candidate lung-resident taxa using a QTL mapping approach. In addition, the two abundant core taxa Lactobacillus and Pelomonas were chosen for independent microbial phenotyping using genus-specific primers. In total, this revealed seven significant loci involving eight bacterial traits. The narrow confidence intervals afforded by the AIL population allowed us to identify several promising candidate genes related to immune and inflammatory responses, cell apoptosis, DNA repair, and lung functioning and disease susceptibility. Interestingly, one genomic region associated with Lactobacillus abundance contains the well-known anti-inflammatory cytokine Il10, which we confirmed through the analysis of Il10 knockout mice. Conclusions Our study provides the first evidence for a role of host genetic variation contributing to variation in the lung microbiota. This was in large part made possible through the careful curation of 16S rRNA gene amplicon data and the incorporation of a QMP-based methods. This approach to evaluating the low biomass lung environment opens new avenues for adva
Genome-wide mapping of gene-microbe interactions in the murine lung microbiota based on quantitative microbial profiling - Animal MicrobiomeBackground Mammalian lungs comprise a complex microbial ecosystem that interacts with host physiology. Previous research demonstrates that the environment significantly contributes to bacterial community structure in the upper and lower respiratory tract. However, the influence of host genetics on the makeup of lung microbiota remains ambiguous, largely due to technical difficulties related to sampling, as well as challenges inherent to investigating low biomass communities. Thus, innovative approaches are warranted to clarify host-microbe interactions in the mammalian lung. Results Here, we aimed to characterize host genomic regions associated with lung bacterial traits in an advanced intercross mouse line (AIL). By performing quantitative microbial profiling (QMP) using the highly precise method of droplet digital PCR (ddPCR), we refined 16S rRNA gene amplicon-based traits to identify and map candidate lung-resident taxa using a QTL mapping approach. In addition, the two abundant core taxa Lactobacillus and Pelomonas were chosen for independent microbial phenotyping using genus-specific primers. In total, this revealed seven significant loci involving eight bacterial traits. The narrow confidence intervals afforded by the AIL population allowed us to identify several promising candidate genes related to immune and inflammatory responses, cell apoptosis, DNA repair, and lung functioning and disease susceptibility. Interestingly, one genomic region associated with Lactobacillus abundance contains the well-known anti-inflammatory cytokine Il10, which we confirmed through the analysis of Il10 knockout mice. Conclusions Our study provides the first evidence for a role of host genetic variation contributing to variation in the lung microbiota. This was in large part made possible through the careful curation of 16S rRNA gene amplicon data and the incorporation of a QMP-based methods. This approach to evaluating the low biomass lung environment opens new avenues for adva
Lire la suite »
 Researchers find possible cause for chemoresistance in bowel cancerLarge quantities of the protein IGF2BP2 not only make bowel cancer grow faster, they also make it resistant to common forms of chemotherapy. This discovery was made by a research team led by Martin Luther University Halle-Wittenberg (MLU) in cooperation with Saarland University.
Researchers find possible cause for chemoresistance in bowel cancerLarge quantities of the protein IGF2BP2 not only make bowel cancer grow faster, they also make it resistant to common forms of chemotherapy. This discovery was made by a research team led by Martin Luther University Halle-Wittenberg (MLU) in cooperation with Saarland University.
Lire la suite »